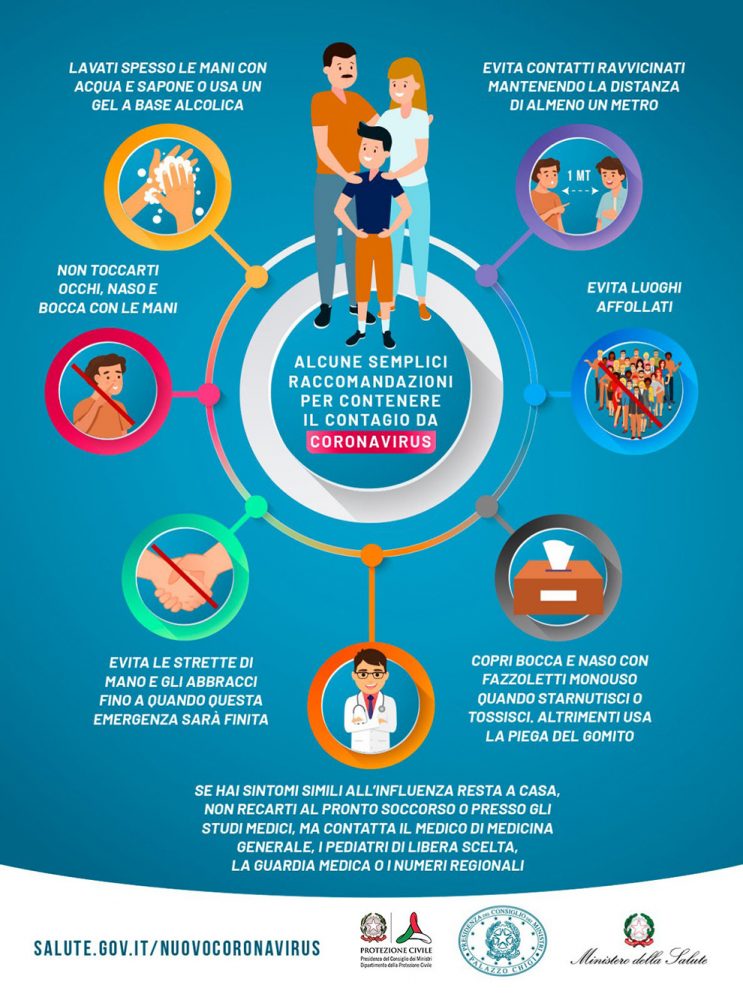
Covid-19, mercoledì webinar ALTEMS Università Cattolica con le associazioni dei pazienti: messaggi per il futuro del Servizio Sanitario nazionale
Mercoledì 3 giugno, ore 16.00, la presentazione online dei risultati dell’indagine nazionale del Patient Advocacy Lab dell’Alta Scuola di Economia e Management dei Servizi sanitari della Cattolica sull’impegno e il ruolo delle associazioni di pazienti durante l’emergenza sanitaria.
Interverranno Sandra Zampa, sottosegretario al Ministero della Salute, Domenico Mantoan, presidente dell’Agenzia Italiana del farmaco (AIFA), Renato Botti, Direzione generale Salute e integrazione sociosanitaria della Regione Lazio.
Roma, 1 giugno 2020 – Sportelli di autoaiuto online, teleconsulti, webinar con gli esperti, raccolta fondi e acquisti agevolati per i presidi sanitari, consegna mascherine e terapie domiciliari, informazione on line contro fake news e timori, webinar di formazione, lezioni di yoga, consigli nutrizionali e supporto psicologico, insieme a molti interventi istituzionali, ossia azioni di advocacy rivolte alle istituzioni o in collaborazione con esse, che hanno portato ad atti normativi a favore dei pazienti, decreti, ordinanze, delibere.
Queste sono solo alcune delle 102 iniziative realizzate da 45 associazioni dei pazienti che nei mesi di marzo e aprile, in piena pandemia da Coronavirus, hanno partecipato all’indagine condotta dall’Alta Scuola di Economia e Management dei Sistemi Sanitari (ALTEMS) dell’Università Cattolica, attraverso il Patient Advocacy Lab (PAL), laboratorio dedicato alle associazioni pazienti. I risultati dell’indagine verranno presentati mercoledì 3 giugno alle ore 16.00 nel webinar dal titolo “Covid-19, iniziative e messaggi per il futuro del SSN dalle associazioni dei pazienti” che potrà essere seguito on line mediante il sito Internet del campus di Roma dell’Ateneo: https://roma.unicatt.it/.
L’incontro sarà aperto da Teresa Petrangolini¸ direttrice del Patient Advocacy Lab dell’ALTEMS, e introdotto da Americo Cicchetti, direttore dell’ALTEMS.
Interverranno Sandra Zampa, sottosegretario al Ministero della Salute, Domenico Mantoan, presidente dell’Agenzia Italiana del farmaco (AIFA), Renato Botti, Direzione generale Salute e integrazione sociosanitaria della Regione Lazio.
Modera Nicola Cerbino¸ capo Ufficio stampa dell’Università Cattolica e della Fondazione Policlinico Universitario Agostino Gemelli IRCCS.
Obiettivo dell’indagine è stato conoscere le attività di advocacy promosse dalle associazioni di pazienti durante l’emergenza Covid-19: le associazioni si sono mobilitate per l’emergenza? quali tipi di azione hanno intrapreso e la loro prevalenza? Quali aree patologiche sono più attive? Ne è nato un quadro variegato e ricco, costruito attraverso la consultazione dei siti web ufficiali delle associazioni che collaborano con il Patient Advocacy Lab (PAL) e grazie alla realizzazione di interviste semi-strutturate con membri delle strutture di governo delle stesse associazioni.
L’area patologica maggiormente rappresentata è quella delle malattie rare (20%), seguita dall’ambito oncologico (18%) e neurologico (13%). Accanto a esse, sono a ogni modo rappresentate numerose altre aree patologiche, a dimostrazione di un impegno generalizzato nel mondo dell’associazionismo. Ogni associazione ha in media condotto 2 azioni anti Covid-19, di cui il 52% riguardano il potenziamento di attività/servizi già erogati prima dell’emergenza, mentre il restante 48% sono servizi nuovi, attivati per far fronte allo stato emergenziale del momento. La maggioranza delle attività (42%) riguardano gli “Interventi istituzionali” presso le autorità sanitarie. A seguire si collocano l’attivazione di web conference e le attività di comunicazione con e per i pazienti. Segue per ampiezza la digitalizzazione dei servizi offerti. A parità di implementazione le attività di realizzazione e consegna mascherine e DPI, e la redazione di documenti di sintesi dei provvedimenti governativi. La formazione a distanza e la raccolta dati completano il quadro, seppure implementate con pochissima frequenza.
Quello che emerge dall’indagine è la varietà delle azioni, con la fantasia e l’innovatività delle iniziative, facilitate da un uso molto diffuso degli strumenti digitali. Forte è stato lo spirito di collaborazione con le istituzioni e delle istituzioni, con un maggior ascolto da parte di quest’ultime delle esigenze dei pazienti, così come le alleanze e il networking tra le associazioni per promuovere azioni comuni. Molti sono i messaggi per il futuro dell’assistenza sanitaria: semplificazione delle procedure, vicinanza e territorio, informazione capillare e personalizzata.
Nel commentare i dati, Americo Cicchetti, direttore dell’ALTEMS ha dichiarato: “E’ evidente come il ruolo delle associazioni si sia rivelato essenziale in questa emergenza e come il nuovo sistema di governance del Servizio Sanitario Nazionale all’indomani del Covid-19 non possa prescindere da meccanismi di integrazione e rappresentanza capaci dare voce a tale soggetto, che esprime, assieme a un punto di vista, anche un bagaglio di competenze e capacità progettuali utili al rinnovamento del modo di fare sanità in Italia”. “Abbiamo voluto raccogliere le testimonianze delle associazioni, offrendo un panorama di un attivismo appassionato e preparato – ha commentato Teresa Petrangolini, direttrice del Patient Advocacy Lab di ALTEMS -. Con il PAL vogliamo svolgere una funzione di counseling e di supporto finalizzata alla crescita di questo mondo associativo, perché il loro operato non sia dimenticato alla fine dell’emergenza, ma possa costituire una comunità di buone pratiche da alimentare, arricchire e far crescere anche in futuro, a beneficio di tutti, cittadini, amministrazioni, operatori sanitari, esponenti politici, aziende private”.
Ufficio Stampa Sede di Roma – ufficio.stampa-rm@unicatt.it
Tel. 06 30154442 – 06 30154295
Sito Internet: www.cattolicanews.it
Social media: @unicatt